Scaffold DIA is an end-to-end software solution for data independent acquisition (DIA) proteomics analysis that provides researchers with the ability to go from raw data to confident protein identification and quantitation. Those familiar with our Scaffold Software will be familiar with the user interface, though Scaffold DIA allows searching raw data directly.
While data independent acquisition shares many similarities with traditional data dependent acquisition (DDA), it requires more time up front to ensure you get good results. For those just starting out, we highly recommend checking out the resources linked below to ensure your first run goes smoothly and you get the best results possible:
This getting started guide is meant to be used with our Scaffold DIA demo data. If following along with your own data you will need:
- A searchable library for the organism in question, to create your reference library
- The corresponding FASTA file
- Reference library DIA data supported by Scaffold DIA
- Experimental DIA data supported by Scaffold DIA workflow option instead.
This guide follows the workflow for creating a reference library, using Prosit and your library data, and searching against it. If you have an existing library and do not need to create one, see the Search Tab section below.
A successful DIA experiment starts with considering three things: windowing schemes, instrument configuration, and choosing the right library. We have a white paper that walks users through setting up your instrument for capturing DIA data, or windowing schemes specifically for Thermo instruments.
As library selection is a critical step, we recommend reading our article on choosing the right library.
Our users have had success using Prosit-derived predicted libraries. We have curated a repository of common model organism Prosit libraries for general use. If your organism is not listed or the collision energies do not match your instrumentation please let us know and we can generate a library for you.
You’ll need to install Scaffold DIA and have an activated license to continue with this guide.
To begin a search, click the New button found on the initial startup Scaffold DIA dialog, the File > New menu option, or the New button on the toolbar. For you first run, most of the search parameters can be left on their default value. Those that should be reviewed are listed below.
You have two options for loading data. If you have an existing library, choose the first option, Search against a FASTA database or existing reference library. If you want to build a chromatogram library and search it all in one step, select the second option, Create a reference library and search against it. For this exercise, select the second option.
Choose a location for your reference library to be saved using the Choose... button.
Scaffold DIA coverts vendor-formatted raw files to internal EncyclopeDIA format (DIA files). These converted EncyclopeDIA files can be searched as if they were raw files, just more quickly. Use the Choose... button next to Save DIA File in... to select a location for your EncyclopeDIA files. Where your raw files are currently stored is a good location to pick.
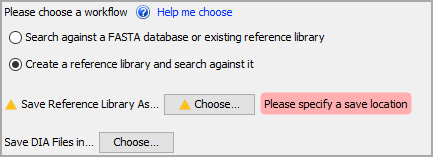
Figure 1. Workflow selection and specifying a save location for EncyclopeDIA files and your library
Expand the Reference Library Creation parameters section to define how the search will be carried out.
Choose a library to search against. Searching a FASTA file directly is discouraged. Libraries here include DDA BLIB libraries, Prosit libraries or DIA ELIB libraries.
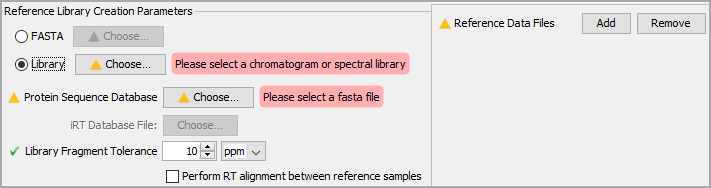
Figure 2. Choose a library, FASTA file and library data
When searching a library directly, you need to provide a FASTA file using the Choose... button next to Protein Sequence Database. This FASTA should be the same one that was used to generate your Prosit library if searching one. Otherwise, choose a FASTA that closely matches the organism in question.
If following along with the demo data, choose the homo_sapiens_prosit_generated_library.dlib for the library and the homo_sapiens_reviewed_uniprot.fasta as the protein sequence database. Also, add the six files found in the library data folder you downloaded.
If you are searching a BLIB file you can choose to provide an iRT database file from Skyline. iRT peptides are not needed to align retention times in Scaffold DIA and thus this step is optional.
Add your library data files using the Add button.
Next, define search parameters specific to the data captured. These options are similar to those found in a traditional DDA search engine and should be familiar to most users.
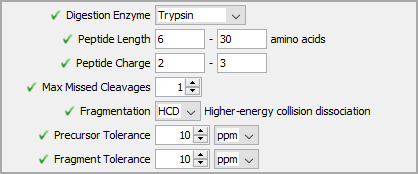
Figure 3. Search parameters similar to those found in a DDA search engine
If following along with the demo data, these can all be left to their default.
When searching a library, only modifications found in the library will be used in the search. When you choose a library, the modifications table will be pre-populated with modifications contained within. Use the Edit... button to correct the name of any modifications in the table.
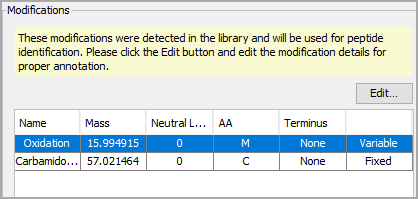
Figure 4. Modifications in the library will be displayed here, Use Edit... to correct the name if needed
Unlike DDA Scaffold, the peptide FDR threshold must be set before the data is loaded. The default is 1% and works well for most runs.
The Data Acquisition Type parameter allows you to define how the data was captured on your instrument. Make sure this matches your LC-MS method as this is a crucial step for processing DIA-MS data. Options here include staggered windows, non-overlapping, or overlapping with margins.
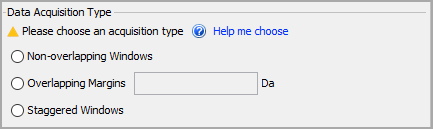
Figure 5. Select the correct data acquisition type
The Precursor Window Size parameter only needs to be defined when loading Agilent data. Otherwise, leave this set to Determine from raw file.
You also have the option to save a workflow file here or load an existing workflow file to use those parameters.
Now close the Reference Library Creation Parameters section and expand the Experimental Data Search Parameters Section.
Parameters in this section are all ported over from the previous section. Simply use the Add button to choose your experimental data.
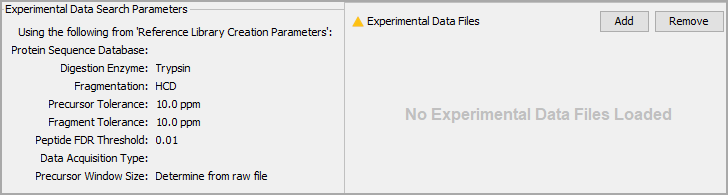
Figure 8. Click add to choose your experimental data
If following along with the demo data, add all of the files found in the Scaffold DIA Demo Experimental Data Part 1 and Part 2.
Analysis Tab
All of the parameters on the Analysis tab can be adjusted after data is loaded. To start with, leave these set to their default values.
Figure 6. Analysis Tab parameters, these can be changed after data is loaded.
Advanced Tab
Start by defining a Processing Directory. Choose a location where you have a lot of disk space. If your computer has a solid state drive selecting this as your processing directory will speed up processing times.
Select the Create a subfolder for intermediate files and retain it option. This will keep the processing files after data is loaded for troubleshooting purposes. Feel free to delete files from older experiments as needed.
Finally, leave the default numbers for quant ions.
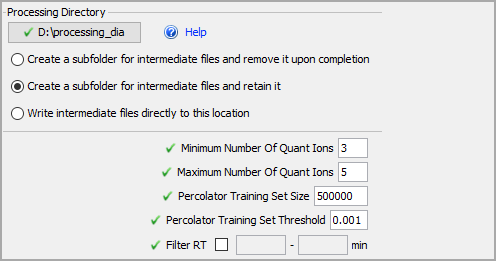
Figure 7. Advanced Tab parameters, select a processing directory and how files should be handled
Once all the yellow triangles are cleared, the loading data process can begin.
To read about how data is displayed in Scaffold DIA, check out Getting Started with Scaffold DIA Part 2.