Scaffold supports loading data from PEAKS in the mzIdentML/MGF file format. The protein and peptide identifications are contained in the MZID file and the spectra are found in the MGF file. While you can load just the MZID file into Scaffold, if you want to see the spectra in the Proteins View, make sure that the MZID and MGF exports are in the same folder. Thankfully, PEAKS has a built-in option for Scaffold that exports both the MZID and MGF file. The following document will explain the process for exporting data from PEAKS. Note, the following screenshots are taken from PEAKS 10.6.
Before exporting, make sure that decoys are visible. This will ensure that they are written to the MZID file. To do so, click on the Preferences icon on the PEAKS toolbar
Figure 1. A portion of the PEAKS toolbar with the Preferences icon highlighted
When the dialog pops up, navigate to the Display Options tab. If the Show Decoy Hits box is not checked, check it now.
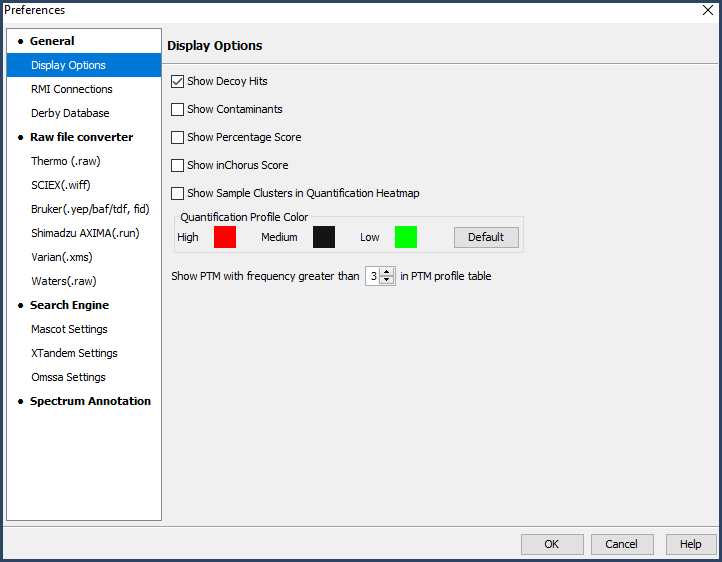
Figure 2. Display options dialog box with the Show Decoy Hits option checked
Set the peptide and protein level score thresholds down to zero. Scaffold's internal probability assignment algorithms work better when they have access to the full set of good and bad PSMs. Providing all of the hits will ensure better probability assignment.

Figure 3. Note the Peptides score threshold and the Proteins score threshold are set to 0
Now, click the Export button. When the Export dialog opens select the For Third Party option. Check the box next to PRIDE/Scaffold. Choose a save location and select Export. This will write both the MZID and MGF file needed to load the data into Scaffold.
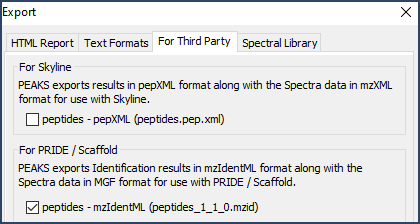
Figure 4. Select the mzIdentML version 1_1_0 export in the for Scaffold section
To load data into Scaffold, create a new experiment and point Scaffold to the exported MZID file. As long as the MGF is in the same folder, the spectra will be visible in Scaffold.
Precursor Intensities
The mzIdentML specification does not allow for precursor intensity information to be stored, thus extra files are needed when loading this data into Scaffold. In order to get intensity information in Scaffold please follow the workflow below:
- Create a folder on your file system for the data to be exported.
- Export the MZID/MGF files according to the workflow above to the folder you created.
- Locate the runInfo folder, this is part of your PEAKS experiment created after you run the search and save the project.
- Copy the entire runInfo folder into your project directory. You now should have a folder with MZID/MGF pairs and the runInfo folder.
- Create a new Scaffold experiment and load the MZID files into Scaffold, making sure to select Precursor Intensity as the Quant type when creating your experiment.