Scaffold can load data from Proteome Discoverer and numerous quantitative options are supported. However, PD must be configured properly to ensure Scaffold has access to both identification and quantitative data.
To begin a few notes:
- Scaffold reads the MSF files created by Proteome Discoverer. For version 2.x both the MSF and the PDStudy file must be in the same directory.
- Make sure to Save All in PD before loading files into Scaffold
- When running the Daemon, Scaffold needs access to the PARAM file instead of the PDStudy file. Make sure this is in the same directory as the MSF files.
Note, at this time Scaffold does not support custom quantitative methods created in PD. Please use the default methods for iTRAQ, TMT or SILAC quantification.
Scaffold also does not support loading one study that was searched using multiple instances of the same search engine node. For example, searching with two Sequest nodes is not supported while searching with one Sequest and one Mascot node is supported.
Prefiltered Mode
Scaffold 4.8.1 introduced prefiltered mode which allows users to bypass Scaffold’s built in probability
model and filter your data using the search engine instead. Use the Percolator node in the processing workflow and set the FDR cutoff to what you consider an acceptable value.
Starting in Scaffold 4.11.0 you will be able to threshold your data using the dropdowns in the Samples view using Percolator-derived probabilities. Otherwise, when you use prefiltered mode you will not be able to adjust the probability thresholds. If using an earlier version of Scaffold it is recommended to then set a custom peptide threshold using the Edit menu.
Quantitative Workflows
Scaffold supports multiple types of quantitative experiments when loading files from Proteome Discoverer including precursor intensity, reporter ion (iTRAQ or TMT), and precursor ion (SILAC) quantitation. Each type of experiment requires different quantitative nodes in the Processing and Consensus workflows. Changes were made for PD 2.2 so workflows are slightly different than those found in earlier versions.
Precursor Intensity Quantitation
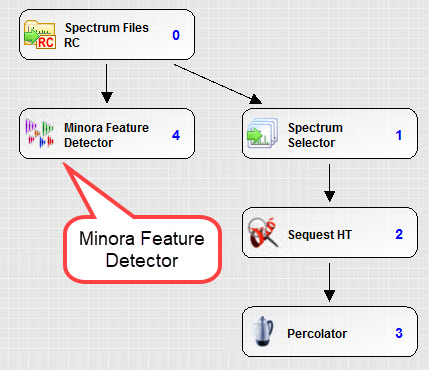
The following is an example processing workflow for precursor intensity quantitation. Note, set the processing parameters such as modifications and tolerances appropriately for the data that was captured.
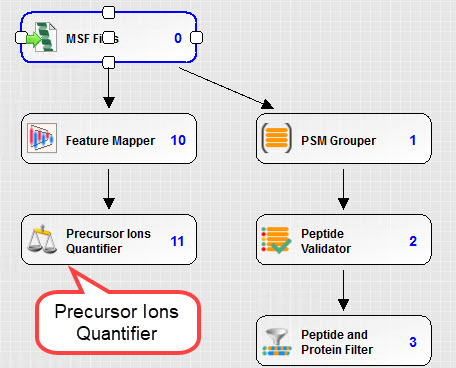
The following is an example consensus workflow for precursor intensity quantitation
SILAC Quantification
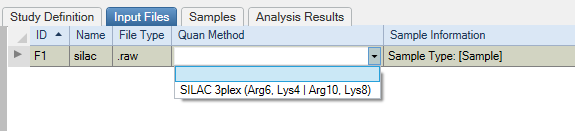
Start by selecting the proper quant method in the Study Definition tab.
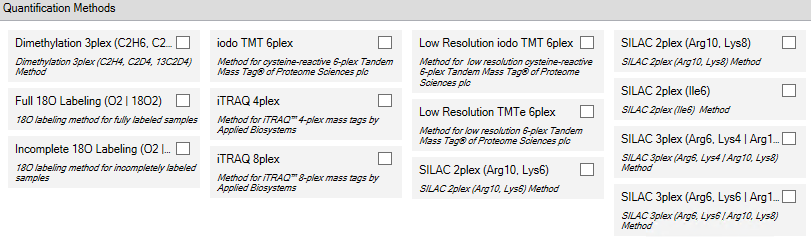
Chose the proper quant method for each file in the Input Files Tab
An example Processing workflow for SILAC data. Make sure to choose the proper quantitative modifications and tolerances in the search engine configuration parameters.

A portion of the Consensus workflow for SILAC quantitation. Note the Precursor Ions Quantifier.
If using the Mascot node as opposed the Sequest HT node, the modifications can be set using the “From Quan Method” dropdown in the Modification Groups section of the search engine parameters. They do not need to be defined as dynamic modifications when this is done.
Reporter Ion Quantification
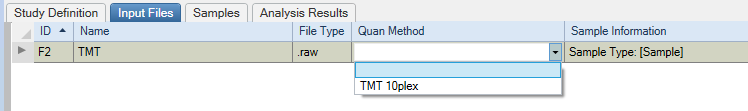
Start by selecting the proper Quant method in the Study Definition tab.
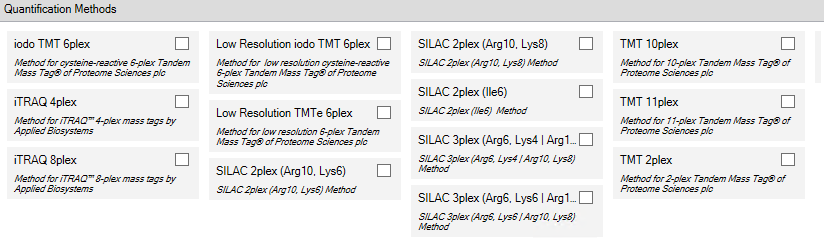
Chose the proper quant method for each file in the Input Files Tab
An example Processing workflow for iTRAQ/TMT data. Make sure to choose the proper quantitative modifications and tolerances in the search engine configuration parameters.

A portion of the Consensus workflow for reporter ion quantitation. Note the Reporter Ions Quantifier.
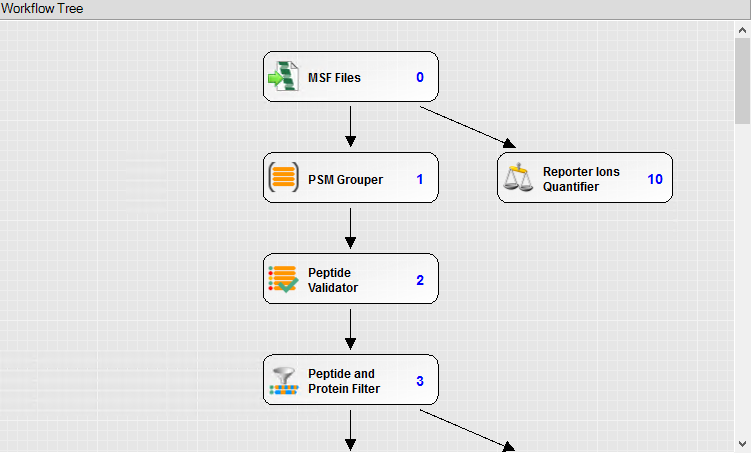
If using the Mascot node as opposed the Sequest HT node, the modifications can be set using the “From Quan Method” dropdown in the Modification Groups section of the search engine parameters. They do not need to be defined as dynamic modifications when this is done.