Introduction
Often researchers receive their processed data in the form of a Scaffold SF3 file from a core facility or collaborator. The following documentation provides a quick-start guide for first time Scaffold Viewer users and provides a short introduction to the data available in the Scaffold file you have received.
Installing the Scaffold Viewer
Installers for Scaffold viewers can be found here, select the proper program, operating system and system architecture (32-bit vs 64-bit) and choose Download Now. Once the file has downloaded double click to run the installer.
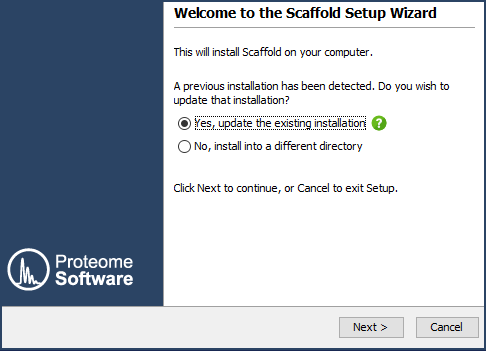
Figure 1. The first page of the Scaffold installer. Click Next > to work through the installer. In most cases the default settings are recommended
Once the installer has completed, you can run Scaffold for the first time. As this guide is primarily intended for users who will run Scaffold in viewer mode only, when prompted for a key by Scaffold, tell the program to continue permanently in viewer mode.
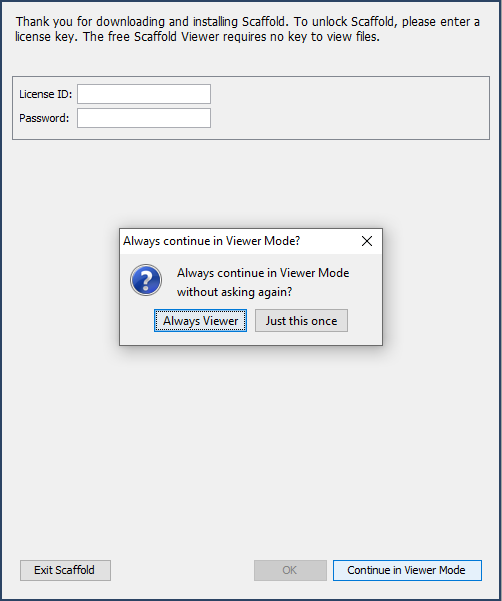
Figure 2. Selecting "Always Viewer" will ensure you are not prompted for a key each time you run Scaffold
Allocating Memory
Scaffold users will see an increase in performance when the program has access to more RAM. Before viewing data for the first time in Scaffold open the Edit > Preferences > Memory tab and allocate about 80% of your systems total RAM (in MB) to Scaffold. Close and reopen Scaffold to save the settings.
Accessing Scaffold Help
This document provides a high-level explanation of features. For answers to detailed questions the Scaffold User Guide is available from the Help menu. Additionally, users will notice blue question mark buttons in various locations around the program. These open an in-program version of the User Guide which can be queried to get quick answers.
The Scaffold File Creation Workflow
The following document goes into detail about the steps involved in creating a Scaffold file. If you received a finished Scaffold file from a core facility these steps will already have been completed.
Scaffold Views
Scaffold focuses predominantly on displaying protein and peptide identifications and quantitative information along with additional metadata such as gene ontology terms via the NCBI or UniProt (see Applying GO Terms). Scaffold is divided into 7 distinct views:
- The Load Data View: Displays the organization of MS Samples into BioSamples along with information relating to search engine parameters, such as fragment and peptide mass tolerance and the searched FASTA database. A Scaffold viewer does not allow users to add additional MS Sample files to a Scaffold file nor can MS Sample files be reorganized into different BioSamples. BioSamples may be organized into categories through the Scaffold Viewer, however.
- The Samples View: This protein-centric display gives information about proteins identified, GO terms, and quantitative values. Peptide and protein level thresholding and filtering can also be done here. The Display Options dropdown menu allows users to toggle between displaying protein identification probability, various quant types, and percent coverage. All of this is given at the experiment level (for all files loaded into Scaffold).
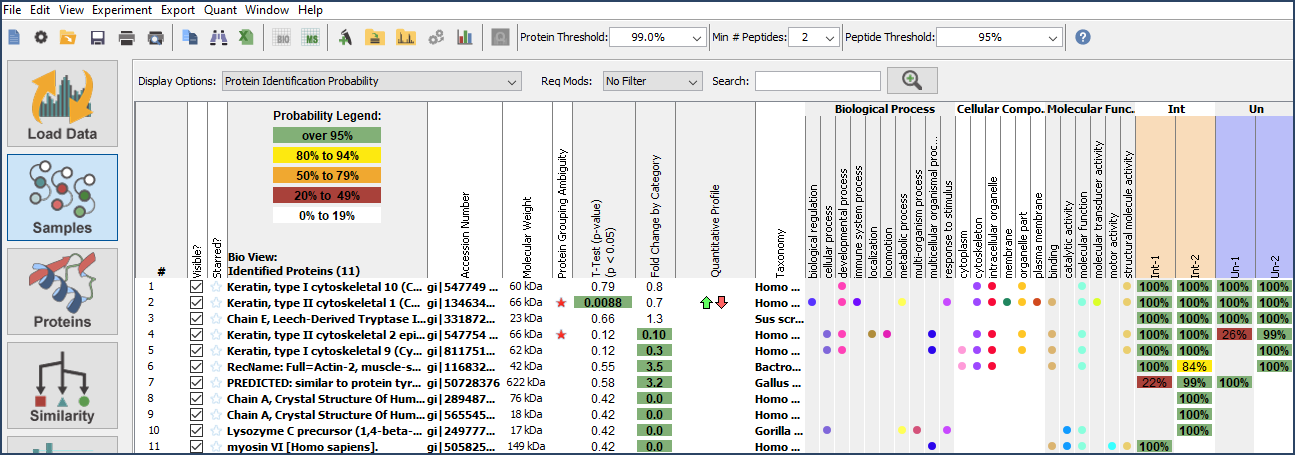
Figure 3. The Samples View complete with GO terms, fold change calculation and T-test added
- The Proteins View: Contains information related to the individual selected protein including the peptides identified (with search engine scores), sequence coverage and spectrum visualizations.
- The Similarity View: Here users can see which peptides are assigned to which proteins, where there are shared peptides and which proteins are excluded from the Scaffold experiment due to the principle of parsimony.
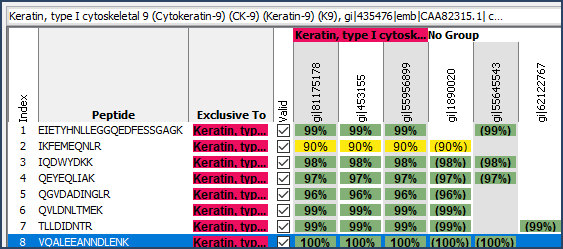
Figure 3. The Similarity View shows how proteins are assembled from the identified peptides
- The Quantify View: Scaffold displays quantitative values based on spectrum counting, or precursor intensity values. This view displays this information at the protein level along with quantitative bar charts, volcano plot, Venn diagram, and GO term charts.
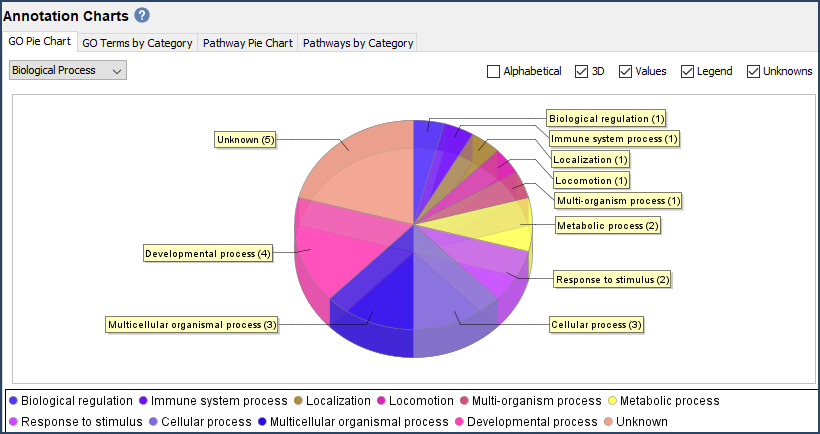
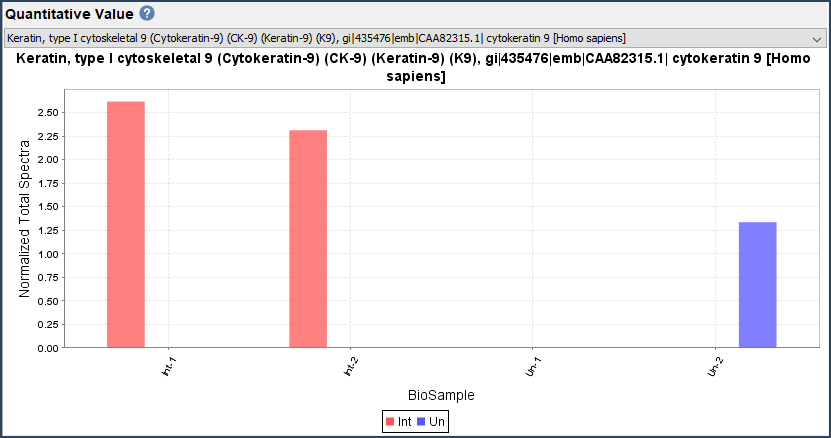
Figure 4. GO Term Pie Chart and Quantitative Bar Chart found in the Quantify View
- The Publish View: Collapses experimental metadata that can be used to begin writing articles for publication in scientific journals.
- The Statistics View: Gives graphical representation of search engine information such as discriminant score histograms, peptide ROC plots, and multi-search engine score scatterplots.
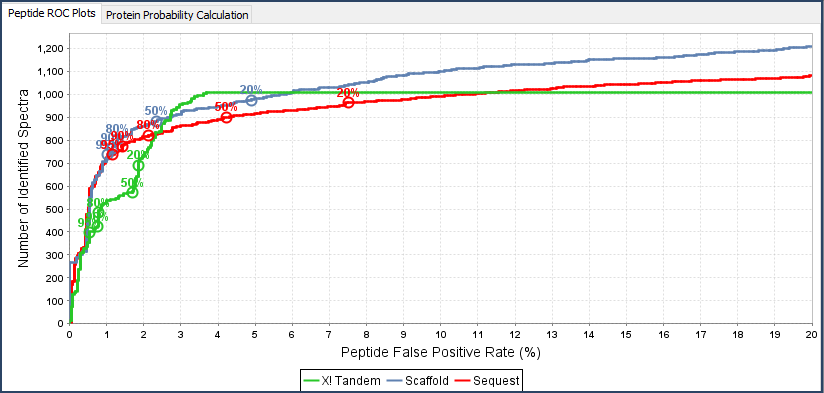
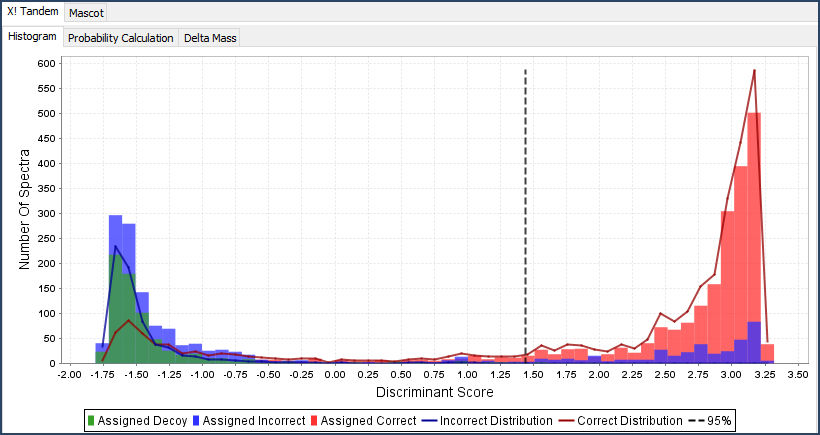
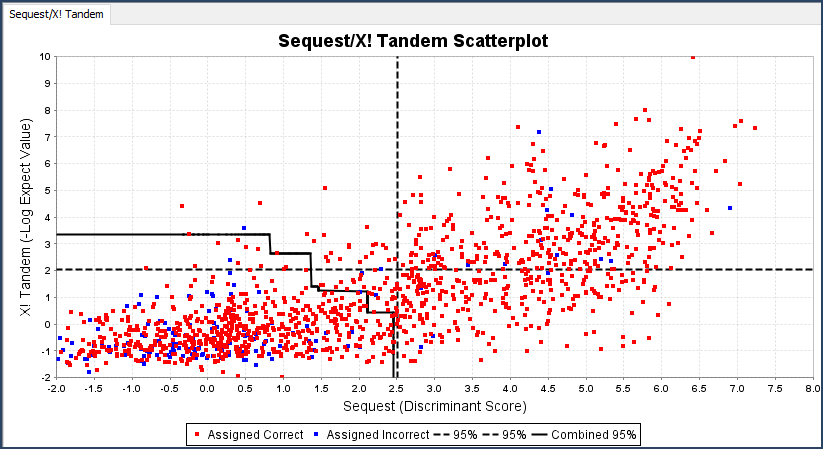
Figure 5. ROC plot, Scatterplot and Histogram available in the Statistics View
Filtering Identifications
Scaffold assigns probabilities to search engine peptide identifications using either Percolator, PeptideProphet, or LFDR and protein identifications using ProteinProphet. This allows users to filter identifications at the peptide and protein level by probability. Additionally, if a decoy search was performed Scaffold allows users to filter by FDR thresholds. Finally, the minimum number of peptides filter allows users to require a minimum number of peptides to be identified.
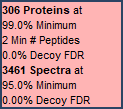

Figure 6. Dropdown menus allow for probability based filtering. Achieved thresholds are displayed in the FDR dashboard found in the lower left corner of the program
Scaffold also provides filtering based on required modifications (using the Req Mods filter) or protein name or accession number (using the Search box). To access more advanced filtering options, select the Advanced Filter button (magnifying glass).

Figure 7. Additional filters are located next to the Display Options dropdown
Scaffold has the ability to add gene ontology terms via two methods. The first involves querying the NCBI GO term database and is built into Scaffold. To do this make sure the NCBI option is selected using the Edit > Edit Annotation Options dialog. Then, the annotations can be added using the Experiment > Add or Edit Annotations... option. Make sure the Go Terms source button is selected and choose the desired option fro the dropdown menu. The second GO term source is the EBI’s GO term knowledge base. Users can add organism specific files to Scaffold and gain GO annotations that way. For detailed instructions see this link.
Scaffold Quantitation
Scaffold allows for quantitative analysis based on spectrum counting (total, exclusive, NSAF, emPAI) or MS1 precursor intensity and allows users to determine differential expression using these quantitative techniques. The Experiment > Quantitative Analysis… dialog box allows users to define the quantitative technique, add statistical tests and fold change calculations. Additionally, this dialog allows users to remove samples from quantitative calculations, toggle inter-sample normalization and define the minimum value used in place of missing values.
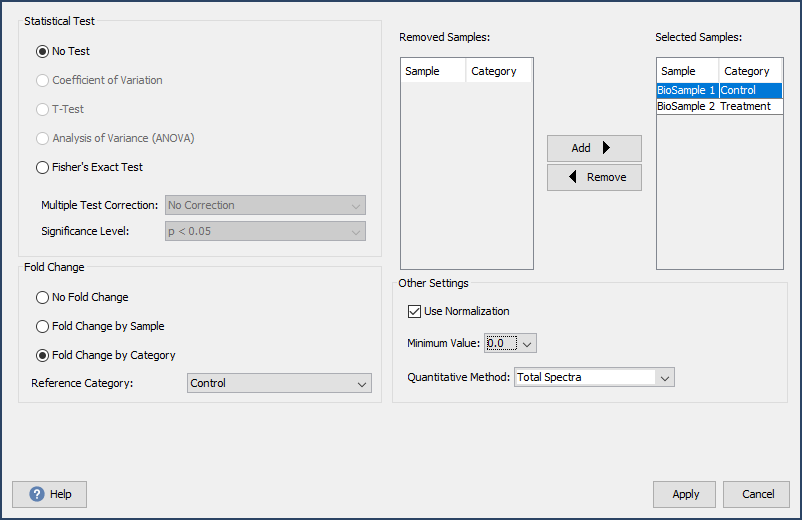
Figure 8. Dialog for adding statistical tests and fold change calculation
Scaffold or Q+S?
If receiving a quantitative experiment, the Q+S button will be active; select this button to enter Scaffold’s quantitative module. Depending on the questions you are trying to answer information may be contained in either Scaffold or Q+S.
Figure 9. The Q+S button allows for select quantitative results to be viewed in the Q+S module
The Q+S module extends the identification information found in Scaffold and provides support for: TMT, iTRAQ, SILAC, and dimethyl based quantitation. Both modules also support MS1 precursor intensity quantitation. Additional quantitative features include kernel density estimation, intensity and error weighting and the ability to organize experiments into categories with proper handling of technical replicates.
Organization of Quantitative Samples
Each experiment must be organized the first time the data is visualized in Q+S, made easy by the Experimental Design Wizard. The wizard is broken into 5 components; simply define a few experimental characteristics and organize samples into reference and non-reference categories. Scaffold extracts quantitative information such as reporter ion or SILAC channel.
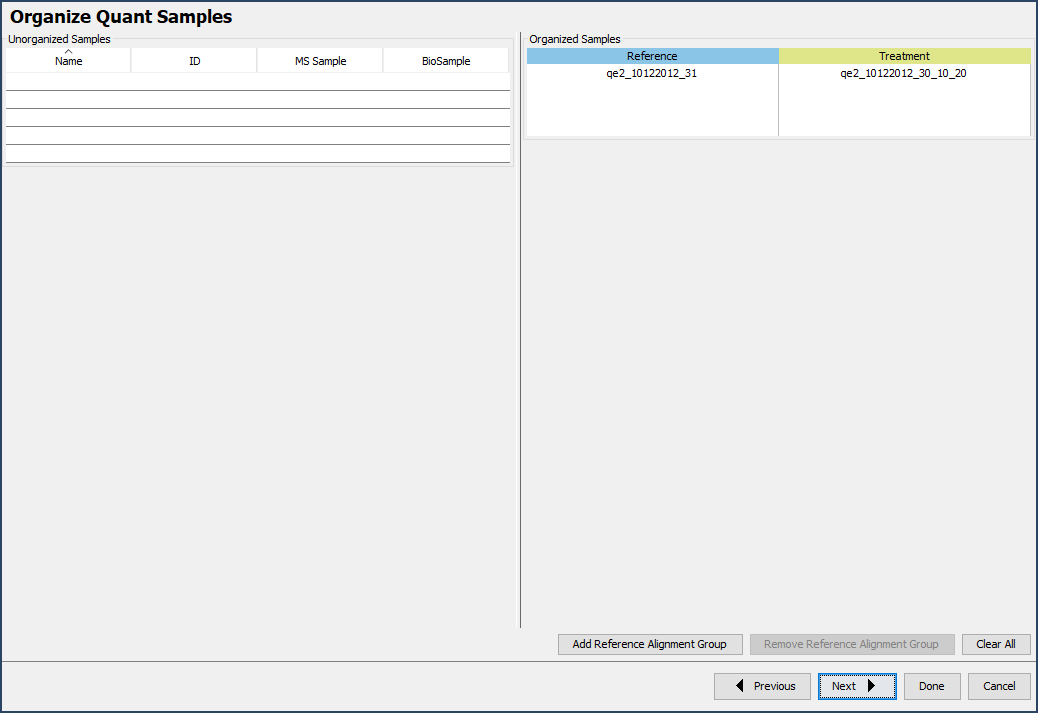
Figure 10. The experimental design wizard, click Next to work through the wizard after defining a few experimental parameters.
The organization of an experiment can be changed after loading into Scaffold Q+S using the Quant > Update Experimental Design… dialog.
Quantitative Testing
The goal of Q+S is to help determine the amount and significance of differential expression. Use the Quantitative Testing dialog found in the Quant menu to add fold a change calculation and statistical test. This dialog gives you options for adding and removing samples from the calculation, applying a multiple test correction such as the Bonferroni or Benjamini-Hochberg correction and gives options for statistical testing based on experimental design.
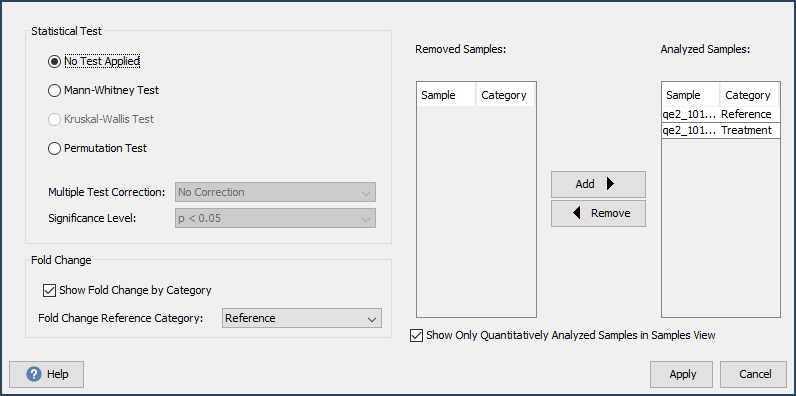
Figure 11. The Scaffold Q+ Quantitative Testing dialog box
Exports
Both Scaffold and the Q+S module give numerous Excel formatted exports, available from the Export menu. These allow users to manipulate data outside of Scaffold and provide data for publication. Additionally, all graphs can be saved by right-clicking.
Additional Resources
Proteome Software has an extensive documentation library, these resources can be found here: