The following document provides introductory information on proteins and the field of proteomics.
- What Are Proteins?
- What Is A Proteome?
- What Is Proteomics?
- Why Care About Proteomics?
- How Are Proteins Identified?
- How Does Mass Spectrometry Identify Proteins?
- How Do Mass Spectrometers Work?
- What Can Proteomics Software Do?
Proteins are one of the four major types of biological molecules. They carry out a wide variety of functions including forming muscle, cellular signaling, turning genes on and off, forming the cellular skeleton, burning energy, catalyzing and inhibiting chemical reactions and controlling the immune system. Proteins do it all. Although every school biology course tells more about proteins than this single web page, this article forms a reasonable introduction.
Protein Structure
Proteins are long chains of amino acids. Twenty common amino acids in various combinations form proteins, so for simplicity, each amino acid is often represented by a letter of the alphabet. Therefore, a protein can be written as a sequence such as MALWMRLLPL. These amino acids are the beginning of the protein insulin. Insulin is a small protein, here is its complete amino acid sequence:
MALWMRLLPL LALLALWGPD PAAAFVNQHL
CGSHLVEALY LVCGERGFFY TPKTRREAED
LQVGQVELGG GPGAGSLQPL ALEGSLQKRG
Many proteins are ten, twenty, or a hundred times bigger than insulin, but all proteins are chains of the same 20 amino acids. There are hundreds of thousands of proteins, each with a different amino acid sequence. What this means is that if you know a protein's sequence of amino acids, you can identify it. This is the basis of protein identification by mass spectrometry.
Within cells, proteins fold these one-dimensional chains into an amazing variety of three-dimensional structures. These 3D structures allow proteins to perform their wide range of functions. The following image shows the multiple levels of a proteins structure.
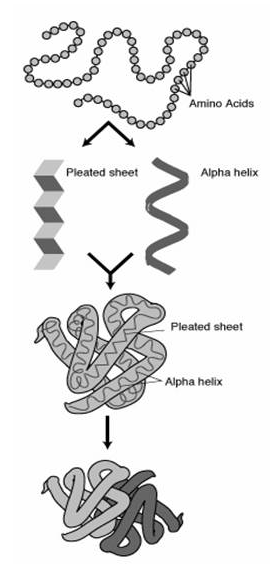
Figure 1. The four levels of structural organization within a protein including 1. Primary: amino acid sequence 2. Secondary: alpha helicies and pleated sheets 3. Tertiary: three-dimensional structure and 4. Quaternary: interaction of multiple sub-units
Much of protein science is devoted to learning these 3D structures and how to predict a protein's function from its structure. A major computational biology problem is predicting the 3D structure from the 1D structure.
Protein Functions
Proteins work together in pathways, sequences of chemical reactions, to perform a wide variety of functions:
- Metabolism: Proteins mediate chemical reactions that use oxygen to burn food for energy. These pathways are complex but well studied.
- Signaling: Hormones are proteins that signal between cells usually in the blood. Proteins also send signal from one nerve cell to another. Signaling pathways, however, are usually interactions among proteins within a cell.
- Regulation: Proteins called enzymes control chemical reactions in cells. Proteins also form gateways in cellular membranes to control what passes into or out of a cell.
- Cellular structure: Proteins define cell shape and form.
- Transportation: Proteins move oxygen, sugar, nutrients and wastes into and around cells. They also move other proteins from where they are made to where they are used.
- Movement: Proteins contract muscles and move cells.
- DNA Transcription: Proteins turn genes on and off.
- Immune System Functions: Special proteins identify germs and other foreign substances and mark them for destruction.
Many proteins are neither rigid nor static. They change shape, open and close, twist and turn. This ability to move lets proteins be tiny machines that can grab and release, push or pull.
Proteins as Catalysts
Unique structures and shape shifting enable proteins to be astoundingly effective catalysts. A catalyst speeds up a chemical reaction. Iron rusts somewhat faster when wet; in other words, water catalyzes rusting. But so what? Inorganic catalysts are dull.
In contrast, enzymes, proteins that act as catalysts, make life possible. Whereas water speeds up rusting only somewhat, enzymes speed up cellular reactions thousands or millions of times. A chemical reaction that takes a fraction of a second with enzymes would take years without it.
A proteome is the sum of all the proteins in an organism, a tissue, or the sample under study. Just as the human genome is the collection of all human genes, so the human proteome is the collection of all human proteins. While your genome is determined exclusively by heredity, your proteome arises from both heredity and environment.
Proteome and Genome
Genes are the blueprints for proteins. Roughly speaking, genes (DNA) make RNA, and RNA makes proteins.

Figure 2. The genomic map of the chicken
The Human Genome Project mapped the human genome, and now genomes for many other species have been mapped. To a first approximation, each gene makes one protein. If you know the genome for a species, then, you know all its proteins. That is, you can predict the amino acid sequence of each protein. Only about one-third of the human proteins predicted by genomics have been isolated and studied. proteomics is not limited to these well-studied proteins, but also deals with the predicted but as of yet unstudied proteins.
Proteome Over Time
The genome of an organism, its DNA, never changes. In contrast, the proteome changes all the time. The amount of each individual protein varies constantly. Protein levels present problems similar to the word problems of grade school arithmetic. Imagine two bathtubs that are filling and draining at the same time.
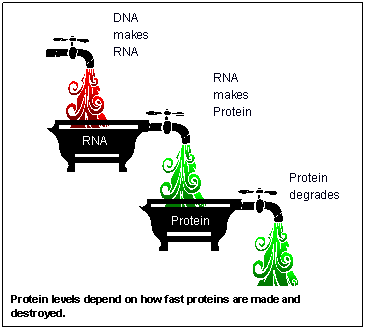
Figure 3. The cascade of DNA to RNA to proteins
The upper tub is RNA. It fills when the gene is turned on so that its DNA makes RNA. It drains as the RNA makes proteins. The lower tub is the protein, which is filled when the RNA makes protein. This tub is drained as the protein degrades. The amount of protein therefore depends on how open each of the three faucets is. To make things more interesting, within living cells these faucets are continuously and independently adjusted.
Also, as the diagram shows, neither the existence of a gene nor the measured levels of RNA corresponds to measured levels of the protein it makes. The gene is turned on at time zero, and RNA builds up. Later, the protein level starts to rise. It may stay high long after the RNA levels fall.
Proteome and Proteins
How many proteins are in the proteome? Because the human genome has about 20,000 genes, you might guess that the human proteome has about 20,000 proteins. But this number is both too big and too small.
Why too small? The classic dogma of genetics is: one gene equals one protein. This is oversimplified; in fact, there are more proteins than genes.
- Genes are made in sections called exons, which can be spliced together in different ways. About half of all human genes have an alternative splicing, with each splice variant corresponding to a different protein.
- Proteins are modified after they are made. Frequently, the proto-protein made from the gene is cut into sections to form many proteins with different properties. For example, the gene CO3_HUMAN can be spliced to form the proteins C3 beta, anaphylatoxin, C3c, C3d, C3f, and C3g — each with different properties and functions.
Why too big? We usually consider the proteome at the level of only one specific organ or body fluid. A human brain cell and a human liver cell each have the same genome of 20,000 genes, but they have very different proteomes, each with only a subset of all possible human proteins.
- Each tissue or bodily fluid has its own proteome. The human body has many proteomes: the blood, liver, brain, and so on. Furthermore, each organ has many types of cells: the brain has various types of neurons and glial cells as well as blood vessels and blood cells.
- Each cell type has its own proteome. Each of these proteomes have only a small fraction of all the possible proteins.
Plasma Proteome
Plasma is blood without the red and white blood cells. One of the best studied proteomes is that of human blood plasma. The plasma proteome has about 1,000 proteins, not 20,000. What's more, almost all the protein by weight is in the most abundant twenty proteins. All other proteins are present in only trace amounts, roughly a billion times less abundant than the common ones. Also, some proteins appear in several splice forms.
Cellular proteomes have more proteins than plasma, but share these features:
- Not every protein is in each proteome.
- Many proteins in the proteome have several forms.
- A small number of proteins make up the bulk of the proteome.
Proteomics is the search for information about proteins. Traditionally, scientists have studied proteins one by one. Proteomics studies a whole proteome at once. Proteomics identifies proteins, modifications of proteins, interactions between proteins, and more.

Figure 4. A genomics laboratory
Proteomics and Genomics
Tens of thousands of scientists have studied proteins in the past century. This effort has resulted in a vast body of knowledge about proteins. Each bit of this knowledge was the result of an immense amount of bench work. A similar situation existed in genetics until gene-sequencing machines were invented and genomics was born. Suddenly, knowledge about the genome exploded. One of the lessons of the Human Genome Project was that a single company with many machines could sequence the human genome. Large-scale biology was born. Proteomics is the application of large-scale biology to protein science. Proteomics is in its infancy, even compared to genomics, which itself is only a handful of decades old.
Proteomics Technologies
Proteomics employs a number of technologies. Indeed, many people define proteomics as the study of proteins using these technologies:
- Mass Spectrometry: The tool that has made proteomics possible. Mass spectrometers are machines for measuring the mass and charge of charged particles.
- Bioinformatics: Software and databases are also enabling technologies for proteomics.
- Liquid Chromatography and 2D Gel Electrophoresis: Ways of separating proteins so that they may be more readily identified.
- Antibodies: People have harnessed the body's immune system to identify proteins. Today, antibodies are the gold standard for protein identification and quantification.
Proteomics Perspectives
Proteomics is the study of all the proteins in a given sample at once. However, within the umbrella of proteomics, proteins can be studied from various perspectives:
- Identification: Simply identifying the proteins in a biological sample turns out to be tricky. Identification is the part of proteome research that has come closest to using techniques of large-scale biology.
- Quantification: It is important for many applications to determine how much of each protein is present in a sample. Differential expression (how protein levels differ between two samples) can be determined by labeling each sample, then comparing them on 1) a 2D gel — a technique called Difference Gel Electrophoresis (DIGE) or 2) on a mass spectrometer — one technique being isotope-coded affinity tagging (ICAT).
- Modification Identification: Proteins are modified in many ways. Frequently a modification changes how the protein works.
- Structures: Function follows form. Determining the three-dimensional structure of a protein helps explain what the protein does, and how it does it.
- Interactions: No protein is an island. Proteins work with other proteins to carry out their functions. To understand their functions, we must understand these interactions.
- Subcellular Locations: Proteins do their work in specific locations within a cell. Proteins interact only with other proteins at the same location.
Proteomics will lead to research breakthroughs allowing doctors to better diagnose and treat diseases. Those pursuing proteomics hope to find biological markers that signal disease, targets for drugs, and detailed understanding of biology on the molecular level. Proteomics may hold the key to personalized medicine
Who Does Proteomics?
People being people, it is not surprising that most proteomics research so far has been aimed at medical applications. This is not to say that you will see proteomics in your doctor's office. Proteomics is a research discipline that is carried out in laboratories at universities, government institutes, biotech firms and pharmaceutical companies.
Proteomics and Nature vs. Nurture
Proteomics has something to offer the long-standing debate about nature versus nurture or heredity versus environment.
The causes of disease exist on a continuum. Some, such as Down syndrome, are completely genetic in origin, while others, such as scurvy, are completely environmental. But most diseases, ulcers for example, are somewhere in the middle: people with different genes will react differently to the same stimuli.
Genomics can contribute most to treating genetic diseases. Because proteins are created from genes, but the amount of a given protein is controlled by the environment, proteomics can shed useful information on the whole spectrum of diseases.
Applications of Research in Proteomics
- Scientific Knowledge: Proteomics has been used to study basic biological questions. For example, what is the structure of the pores between the nucleus and the cytoplasm of a cell?
- Diagnosis: Proteomics has looked at tumor biopsies and blood samples for indicators of cancer, birth defects, and other medical conditions. One diagnostic challenge is distinguishing between disorders with similar symptoms, but requiring different treatments.
- Monitoring: Proteomics can also search for biomarkers that indicate the stage of a disease, or the response of the patient to treatment.
- Drug Discovery: Most drugs target proteins, so it makes sense to use proteomic techniques to search for drug candidates.
Diagnosis Using Multiple Proteins
Some clinical tests look for an elevated level of a single protein, signaling a specific disease. Proteomics can be used to search for these biological markers for disease. Unfortunately, most diseases are not easy to diagnose with a single protein. Cancer researchers are now exploring an alternative approach: looking for a pattern of many different proteins. They hope that even though no single protein indicates the disease, the expression pattern of dozens of proteins may.
Proteomics and Personalized Medicine
Different people have different responses to treatment:
- Some are cured.
- Some don't respond.
- Some experience a bad side effect.
The goal of personalized medicine is to test patients to determine which medicine will work for them without unpleasant side effects. To reach this goal, research is needed. Proteomics will fuel this drive toward niche drugs and targeted therapies.
Proteomics Beyond Medicine
Proteomics is also being applied to environmental and agricultural problems. For example, proteomics is being used for studying food safety, the environmental effects of wheat development, sleep, and for fighting bioterrorists.
The Lighter Side of Proteomics
Protein scientists have also applied their techniques to study the foam in a glass of beer, sexual wanderings, what mosquito's sniff, and reindeer antlers.
Today, medical clinics and research laboratories routinely identify proteins using antibodies. This requires technicians to have an antibody for each protein they wish to identify. But so far, we’ve identified antibodies for only a limited set of proteins. An alternative identification technique, recently perfected, uses mass spectrometry.
Strategies For Identifying Proteins
Proteins have both a one- and three-dimensional structure:
- A protein can be identified by its one-dimensional structure or amino acid sequence. This is the strategy used by mass spectrometer-based techniques that form the bulk of today's proteomics. (There's more on this below.)
- Proteins can also be identified by their three-dimensional structures. Antibodies have a 3D shape that fits the shape of a protein, somewhat like puzzle pieces fit together.
Antibodies- The Once and Future Proteomics King
In clinical practice today, proteins are always identified by antibodies. This has both good and bad aspects:
How is antibody identification good?: An antibody locks on to a specific protein even when surrounded by thousands of others. So antibodies don't require proteins to be separated, as mass spectrometers do. Antibodies are also sensitive: they can detect minute amounts of a protein. For many proteins, you can buy an antibody from a catalog.
How is antibody identification limited?: Today, antibodies are not available for all proteins and those that are available are costly ($200 - $300 per 50 micrograms). Finally, you must know precisely which protein you are looking for, so that you can get the correct antibody. But proteomics experiments frequently seek to identify unknown proteins. Today, therefore, antibodies are used in proteomics only as a confirmatory step, after mass spectrometry has identified the proteins.
Microarrays are tools that allow researchers to measure the expression of a great many genes simultaneously. Their success has led some to suggest that antibodies be arranged in protein arrays, a panel of antibodies to detect a large number of proteins simultaneously. Some protein arrays are available commercially, but they are still very limited. Most forecasts say that protein arrays covering a large percentage of proteins will be important for proteomics someday, their use is not widespread yet.
Mass Spectrometry Strategies for Identifying Proteins
Mass spectrometry can identify proteins via multiple strategies:
- Tandem mass spectrometry (abbreviated MS/MS) is currently the most popular technique in research labs. Tandem mass spectrometry uses two mass spectrometers hooked together to analyze peptides, shorter sequences of amino acids that make up proteins. The first machine weighs a peptide, the second identifies it.
- Protein Mass Fingerprinting (PMF) is a somewhat older technology that uses a single mass spectrometer; it isn't as accurate at identifying proteins as MS/MS.
- Intact protein mass spectrometry looks at proteins rather than peptides. This top down approach has been successful with the high accuracy that is now possible with the very expensive Fourier transform mass spectrometer.
All of these mass spectrometry strategies work only on samples containing just a few different proteins. Since biological samples have hundreds or thousands of proteins, the proteins in these biological samples must be separated out into simpler samples.
Protein Separation
Cells and biological fluids such as blood have hundreds or thousands of proteins. Most protein identification methods, however this makes protein identification difficult. The mass spectrometry strategies discussed above, for example, all require separating the proteins first. Protein scientists have spent years developing techniques for separating the proteins in a complex mixture. A good part of the lab work in proteomics consists of applying these techniques.
Electrophoresis is the separation of proteins by applying an electrical current to proteins in a gel. One-dimensional gels, can depended upon pH or on the protein's mass. In 2-D gels, proteins are separated by both mass and pH.
Protein Separation: High Performance Liquid Chromatography (HPLC)
High Performance Liquid Chromatography (HPLC) separates proteins as they pass through a column packed with small particles that form a porous matrix. The proteins are suspended in a solution that flows through the column. The proteins stick to the particles in the column to some degree, but they eventually wash out.
Because different proteins have different chemical properties, they wash out at different times. By exploiting these different chemical properties, proteins can be separated in several ways:
- Ion exchange, specifically Strong Cation Exchange (SCX), separates proteins based on their pH.
- Reverse phase separates proteins based upon their hydrophobicity, how much the protein attracts or repels water.
- Size exclusion separates proteins based on their size.
- Bio-affinity separates proteins based on the degree to which proteins stick to antibodies or other substrates.
In most experiments, several of these separation techniques are combined to separate the proteins in each biological sample. For example, one common method is to:
- Concentrate the proteins with a bio-affinity HPLC or size exclusion HPLC.
- Separate the proteins on a 2-D gel.
- Digest proteins into peptides.
- Separate the the peptides with a reverse-phase HPLC.
Another method (sometimes known as MudPIT or shotgun proteomics) is to:
- Digest proteins into peptides.
- Separate the peptides with ion-exchange HPLC.
- Separate the peptides further with reverse phase HPLC
How Does Mass Spectrometry Identify Proteins?
Mass spectrometers identify proteins in a manner analogous to using fingerprints to identify a person. Each protein has a mass spectrum with unique characteristics. To minimize confusion, proteins must first be separated from protein mixtures; then they can be examined with a mass spectrometer.
The most common form of protein identification with mass spectrometry uses a tandem mass spectrometer (abbreviated MS/MS). As explained below, identifying a protein with MS/MS analogous to identifying a person using fingerprints.
Clarification: In proteomics, the term Protein Mass Fingerprinting (PMF) describes a protein identification procedure using a simpler mass spectrometer. The broader analogy of MS/MS to fingerprinting is used here only to explain the concepts.
| Fingerprint Identification | Protein Identification |
|
Fingerprints at a crime scene might be mixed in with hundreds of other fingerprints. The first step is to collect and separate the fingerprints. The common way to separate fingerprints is to look at different surfaces. If they overlap, fluorescence can sometimes separate them. |
A biological sample contains hundreds or thousands of proteins, all mixed together. The first step is to separate them. One common technique uses two-dimensional gels. The spots on the gel are different proteins, separated horizontally by pH and vertically by mass (the two dimensions). |
| Fingerprints are identified by looking at each finger — that is, the hand print at the top of the page is considered as the four individual prints below. A protein's peptides are analogous to these individual fingers. While each person has only 4 fingers, one protein may have 18 peptides, another 27 peptides and yet another only 6 peptides. |
Mass spectrometers can identify proteins better if they are first broken down into tryptic peptides. Tryptic peptides are chains of amino acids that occur when the proteins are digested with the enzyme trypsin. The tryptic peptides for hemoglobin are shown here: VHLTPEEK, SAVTALWGK, VNVDEVGGEALGR, LLVVYPWTQR FFESFGDLSTPDAVMGNPK, VK, AHGK, K, VLGAFSDGLAHLDNLK, GTFATLSELHCDK, LHVDPENFR, LLGNVLVCVLAHHFGK, EFTPPVQAAYQK, VVAGVANALAHK |
| The fingerprints have to be further sorted out by whether they have a whorl, a loop, or arch before they can be identified. | The peptides have to be further sorted before they can be identified. This is often done in the following two steps:
|
| Now the individual fingerprints are examined in detail. Certain identifying features are located, such as a core (the center of a loop) and a delta (a place where ridges divide). | Now the second part of the tandem mass spectrometer examines the individual peptides in detail for identifying features — the distances between the peaks in the mass spectral graph. The peaks represent the number of ions in the peptide's spectrum that have a certain mass and charge. |
| A person is identified by matching the identifying features of the fingerprints to a database of fingerprints. An identification is more believable if it is based on matching fingerprints from several fingers. |
A protein is identified by matching the identifying features of the peptides to a database of proteins. An identification is more believable if it is based on matching mass spectra from several peptides. |
How Do Mass Spectrometers Work?
Mass spectrometers measure the mass of charged molecules. A charged molecule moves through an electrical or magnetic field in a precise way determined by its mass. Mass spectrometry became important to proteomics when researchers discovered how to gently put a charge on proteins without destroying them.
Tandem Mass Spectrometry
Tandem mass spectrometers are built out of two mass spectrometers. The first one selects the peptides by mass one by one and the second mass spectrometer reads out the intensities of the fragment ions of each peptide.
Many different combinations of mass spectrometers have been tried. A popular configuration today is a quadrupole linked to a time-of-flight or Q-TOF. Another common combination is one time-of-flight linked to another time-of-flight a TOF-TOF.
Ion-trap mass spectrometers are unique in that they can trap the peptide ions. The ion trap holds the peptides until they fragment, then it switches mode and measures the fragment ions. Thus the ion trap acts as both the first and second mass spectrometer. Because this is the functional equivalent of a tandem mass spectrometer for the price of a single one, ion traps are popular with those who have limited budgets.
Mass spectrometers for proteomics are expensive. A used ion trap costs $50,000 to $100,000. A new Q-TOF or TOF-TOF costs $300,000 to $500,000. A new FTICR costs around $800,000.
Tandem Mass Spectrometer Components
To identify proteins using a tandem mass spectrometer, the instrument needs:
- a way to ionize the peptides
- a first mass spectrometer that selects a peptide by its mass
- a means of fragmenting this peptide
- a second mass spectrometer to read out the fragments
Mass Spectrometry Principles
A mass spectrometer exploits either of two basic physical principles:
- Charged particles curve when traveling though a magnetic field.
- Heavier particles accelerate more slowly.
Many different mass spectrometry designs: quadrupole, ion-trap, Fourier transform ion-cyclotron resonance use magnets.
Time-of-flight mass spectrometers measure the time it takes a given peptide to travel a given distance.
Ionization of Peptides
Proteins in living organisms are in liquid and are usually not charged. Mass spectrometers can measure only charged particles in a gas. Mass spectrometry for proteomics became possible only when this problem was solved. Two people received Nobel prizes for their solutions:
- One solution is electrospray ionization (ESI). Peptides in an acidic liquid are sprayed in a fine mist through a series of capillaries to separate the ions from the solvent. The liquid in the droplets evaporates, leaving the peptide plus positively charged hydrogen ions from the acid to enter the mass analyzer. In some instruments, a cloud of nitrogen gas will pass through the spray, causing desolvation.
- The second solution is Matrix-Assisted Laser Dissociation Ionization (MALDI). Peptides are dried onto a solid surface, the matrix, which is then hit with a laser beam. The peptides float free as the matrix disintegrates.
Fragmenting Peptides
After being selected in the first component of the tandem MS/MS, the peptides are dissociated into fragments that the second component can analyze. The detectors in a mass spectrometer count charged particles, ions. To be detectable, therefore, the fragments must have an associated charged hydrogen ion, a proton. This mobile proton determines the peaks in the mass spectrum. You can try fragmenting proteins yourself (in cyberspace) with MS-Product, a program that predicts the way that peptides fragment.
Interpreting Spectra of Fragmented Peptides
Peptides are most likely to break at their weakest link. Mass spectrometers are tuned so that the weakest link is between the amino acids. When the peptide breaks, both ends can be detected (if charged), so each break gives two daughter ions. These daughter ions form the MS/MS spectrum.
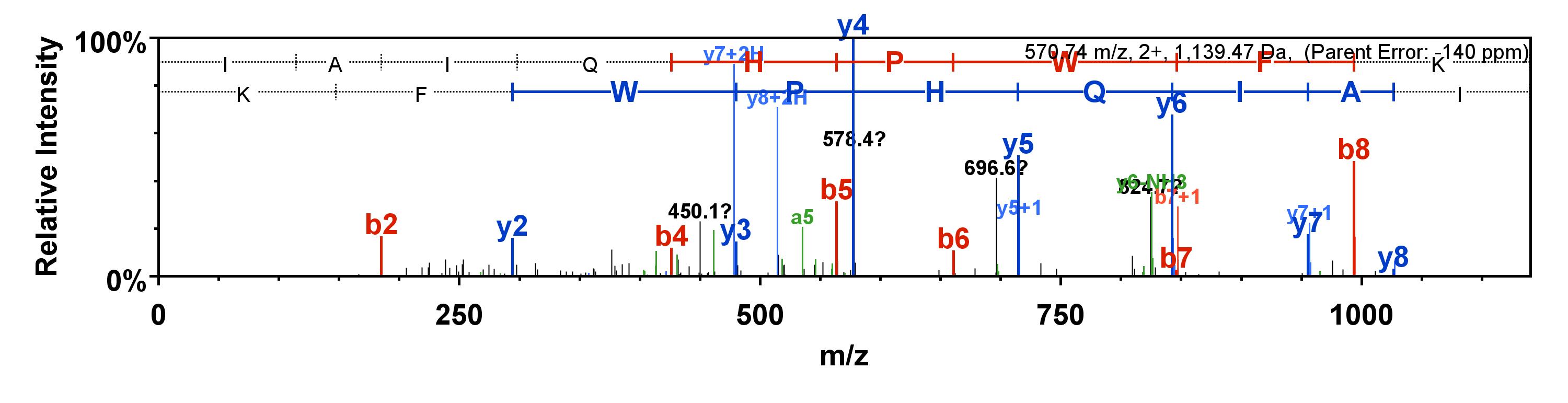
Figure 5. An example MS/MS spectrum
In the figure above, the peptide consists of the amino acid chain IAIQHPWFK. If it fragments perfectly between the amino acids, it will create the daughter b-ions on the left (b1=I, b2=IA, b3=IAI …) and the daughter y-ions on the right (y1=K, y2=FK, y3=WFK…). These daughter ions yield the MS/MS spectrum seen above.
What Can Proteomics Software Do?
Bioinformatics applies software applications and databases to genomics and proteomics. Protein databases are populated with the results of classical protein research, as well as predictions computed from genomics. Bioinformatics software controls instruments and analyzes the results.
Proteomics Databases
Protein databases are at the heart of proteomics. These databases are usually freely available to everyone. The National Center for Biotechnology Information (NCBI) maintains a number of these databases. Additionally, the UniProt KB is an excellent source of FASTA protein databases. The Molecular Information Agent and DBGET are other graphical portals showing interconnected protein and gene databases. A more comprehesive list of specialized proteomic and genomic databases is published each year by Nucleic Acids Research along with articles describing each one.
Varieties of Protein Databases
Database information can be located from numerous sources online:
- General information about proteins: The most famous ones are SWISS-PROT, IPI, PIR, and NCBI. These are combined in UniProt, the world's most comprehensive catalog of information about proteins. A search on the Bioinformatic Harvester returns all that is known about a protein and its related gene in fifteen different databases.
- Protein sequence databases: Amino acid sequences in the FASTA format are used with mass spectrometry to identify proteins. These databases are often a portion of the general protein databases listed above, SWISS-PROT, IPI, PIR, NCBI and UniProt.
- Proteomics databases: Data collected in proteomics experiments such as the protein identifications in PeptideAtlas, PRIDE, the Open Proteomics Database, and the Global Proteome Machine. There are also databases of experimental 2D gels.
- Three-dimensional structures of proteins: The most well-known is PDB.
- Protein-protein interactions: Which proteins interact and with whom.
- Gene Ontology: A database of terms that classify protein functions, processes and subcellular locations. The NCBI and UniProt are two excellent resources for GO terms.
- Databases that relate proteins and genes to diseases: The most well-known is OMIM.
- Pubmed: The database of abstracts of all journal articles relevant to biology, medicine, and proteomics.
Bioinformatics Programs
Bioinformatics software for proteomics has three main parts:
- Interpreting mass spectra: Identifies proteins (discussed in more depth below).
- Computational biology: Largely aimed at computing the three-dimensional shape of a protein from its one-dimensional amino acid sequence. A protein's 3D structure determines its function.
- Sequence comparison: Seeks to understand how proteins work by seeing what other proteins are similar to them. The standard program comparing sequences is BLAST or variations such as PSI-BLAST. For each protein in the NCBI database, BLink lists all other proteins matched by a pre-computed BLAST.
Protein Identification Software
As the diagram above shows, protein identification using tandem mass spectrometry requires software at a number of steps.
- Instrument control software runs the tandem mass spectrometer. In particular, it decides when a peak from the first component is good enough to examine in detail, and then switches to the second component to analyze the peptide fragments. Dynamic exclusion is the technique that prevents the second component from examining the same thing repeatedly.
- Peak-finding software takes the raw data from the tandem mass spectrometer and outputs a peak list. Peaks represent the number of peptides of a given mass and charge.
- Search engines search the spectra of peaks against a protein sequence (FASTA) database. SEQUEST and Mascot are the leading commercial search engines. SEQUEST Sorcerer is a hardware accelerated version of SEQUEST. You can try Mascot on the web. If you need test data to try with Mascot, the Peptide Atlas data repository has publicly available proreomic data sets. There are numerous open source search engines such as X! Tandem and Maxquant.
- An alternative to searching the spectra in a protein sequence database, is to first determine from each spectrum what the amino acid sequence could have produced this spectrum. Then search with this sequence against the FASTA database. This approach is called de novo sequencing. Some de novo programs are: PEAKS, a commercially supported program, Lutefisk, an open source program and PepNovo, an web based program.
- Yet another approach is to find sequences of three to five amino acids using partial de novo sequencing, which is simpler than de novo sequencing the whole peptide. Then you can search the FASTA database with these sequence tags. InsPecT is a web based tag search program.
- Each search alternative identifies proteins, but with some degree of uncertainty. These results can be improved by using statistics to maximize the number of proteins identified while minimizing the false identifications. This analysis is available with Proteome Software's Scaffold program.